Cardiomiopatia Hipertrófica: O Inimigo Silencioso que Mora no Coração
SAÚDE E BEM-ESTAR
5/28/202617 min ler
A doença genética mais comum do músculo cardíaco afeta milhões de pessoas no mundo — a maioria sem saber. Entenda o que ela é, como age, quem ameaça e o que a ciência já sabe sobre como enfrentá-la.
O coração que cresce demais
Imagine um motor que, em vez de trabalhar de forma eficiente, começa a engrossar suas próprias paredes, tornando-se progressivamente mais rígido, mais denso, mais difícil de operar. Esse motor não para de bater — mas o faz com cada vez menos eficácia, e às vezes, de maneira perigosamente caótica. Essa é, em termos simples, a realidade vivida por quem carrega no peito a cardiomiopatia hipertrófica, uma das doenças cardíacas genéticas mais prevalentes do mundo e, paradoxalmente, uma das menos conhecidas pela população geral.
A cardiomiopatia hipertrófica — conhecida pela sigla CMH — é uma condição em que o músculo cardíaco, o miocárdio, se torna anormalmente espessado, ou seja, hipertrofiado, sem que haja uma causa secundária óbvia para isso, como hipertensão arterial grave ou doença valvar. Ela não é resultado de um estilo de vida desregrado, não é causada por sedentarismo ou má alimentação, e tampouco nasce de uma infecção ou trauma. Ela vem de dentro — codificada nos genes, transmitida de geração em geração, silenciosa por anos a fio, e capaz de se revelar de maneira abrupta e devastadora.
Segundo dados da literatura médica mundial, estima-se que a CMH acometa aproximadamente 1 em cada 500 pessoas na população geral. Isso a torna, sob qualquer perspectiva, uma doença comum — embora raramente seja tratada com essa urgência nos consultórios de atenção primária. No Brasil, com uma população superior a 210 milhões de habitantes, esse índice representa potencialmente mais de 400.000 pessoas convivendo com a condição, com graus variados de comprometimento. Uma parcela considerável dessas pessoas nunca recebeu o diagnóstico.
Nasceu assim: a origem genética da doença
Uma das perguntas mais frequentes feitas por pacientes e familiares ao ouvir o diagnóstico de cardiomiopatia hipertrófica é: "Mas de onde veio isso?" A resposta, embora tecnicamente complexa, pode ser dada de forma direta: em grande parte dos casos, veio do DNA.
A CMH é, em sua essência, uma doença genética. A forma mais comum é herdada de maneira autossômica dominante, o que significa que basta um único gene alterado — recebido de um dos pais — para que a pessoa desenvolva a condição. Cada filho de um portador tem 50% de chance de herdar a mutação. Não há distinção de sexo: homens e mulheres são igualmente susceptíveis à herança genética, embora a expressão clínica da doença — ou seja, como ela se manifesta — possa diferir entre os gêneros.
As mutações mais frequentemente associadas à CMH afetam genes que codificam proteínas do sarcômero, a unidade funcional básica do músculo cardíaco. O sarcômero é a estrutura responsável pela contração muscular: é ali que as fibras de actina e miosina deslizam umas sobre as outras para gerar o movimento que chamamos de batimento cardíaco. Quando um gene que codifica uma dessas proteínas sofre mutação, toda a mecânica da contração é alterada.
Os genes mais comumente envolvidos são o MYH7, que codifica a cadeia pesada da beta-miosina, e o MYBPC3, que codifica a proteína C ligante de miosina. Juntos, esses dois genes são responsáveis por mais da metade de todos os casos identificados geneticamente. Mas a lista não para aí: mutações nos genes que codificam troponina T cardíaca (TNNT2), troponina I (TNNI3), tropomiosina alfa-1 (TPM1), actina cardíaca (ACTC1) e outras proteínas sarcoméricas também figuram no espectro da doença.
O que torna essa herança ainda mais complexa é o fenômeno da penetrância variável e da expressividade variável. Isso quer dizer que duas pessoas com a mesma mutação genética — inclusive membros da mesma família — podem ter manifestações clínicas completamente diferentes. Uma pode ter espessamento moderado do ventrículo esquerdo e levar uma vida praticamente normal. Outra pode desenvolver obstrução grave do fluxo sanguíneo, arritmias perigosas e insuficiência cardíaca severa. Por que isso acontece? A ciência ainda não tem todas as respostas. Fatores modificadores genéticos e ambientais provavelmente influenciam o curso da doença, mas o mapeamento completo dessas interações permanece como uma das fronteiras mais ativas da pesquisa cardiológica.
Vale ressaltar que nem todos os casos de CMH são herdados. Uma fração menor dos pacientes — estimada entre 5% e 10%, a depender da série estudada — apresenta mutações de novo, ou seja, alterações genéticas que surgem pela primeira vez naquele indivíduo, sem que os pais sejam portadores. Nesses casos, não há histórico familiar, o que torna o diagnóstico ainda mais desafiador e frequentemente tardio.
O que acontece dentro do coração
Para entender os estragos que a CMH provoca, é necessário primeiro compreender o que normalmente acontece no interior de um coração saudável.
O coração é dividido em quatro câmaras: dois átrios (superiores) e dois ventrículos (inferiores). O ventrículo esquerdo é a câmara principal de bombeamento — é ele que recebe o sangue oxigenado proveniente dos pulmões e o ejeta com força para a aorta, de onde o sangue seguirá para todo o organismo. Para funcionar com eficiência, as paredes do ventrículo esquerdo precisam ter uma espessura adequada, geralmente entre 6 e 12 milímetros em adultos, e devem ser capazes de relaxar completamente durante o enchimento e se contrair de forma potente durante a ejeção.
Na cardiomiopatia hipertrófica, esse equilíbrio é rompido. As paredes do ventrículo esquerdo se espessam de forma anormal — em casos moderados, chegam a 15 ou 16 milímetros; em casos graves, podem ultrapassar os 30 milímetros, o que representa mais do que o dobro da espessura normal. O espessamento é frequentemente assimétrico, afetando de maneira desproporcional o septo interventricular, a parede que separa os dois ventrículos.
Esse espessamento excessivo gera uma cascata de problemas mecânicos e elétricos:
Disfunção diastólica: Com as paredes mais espessas e rígidas, o ventrículo perde a capacidade de se relaxar adequadamente. Isso compromete o enchimento da câmara entre os batimentos, reduzindo o volume de sangue que entra e, consequentemente, o que é ejetado para o organismo. O resultado é sensação de cansaço, falta de ar e intolerância ao esforço.
Obstrução da via de saída: Em aproximadamente dois terços dos pacientes com CMH, o espessamento do septo interventricular provoca uma obstrução dinâmica — variável, que piora com o exercício — no caminho que o sangue percorre para sair do ventrículo esquerdo em direção à aorta. Essa obstrução é frequentemente agravada por um movimento anormal da valva mitral (o movimento anterior sistólico da valva mitral, ou SAM), que passa a ser atraída para a via de saída durante a contração, bloqueando ainda mais o fluxo. O coração precisa trabalhar sob pressões progressivamente maiores para vencer esse obstáculo, o que acelera o remodelamento patológico.
Isquemia miocárdica: O músculo cardíaco hipertrofiado tem maior demanda de oxigênio, mas o suprimento sanguíneo pelos vasos coronarianos intramurais (menores artérias dentro do próprio músculo) frequentemente está comprometido. Essa desproporção entre oferta e demanda gera episódios de isquemia — falta de oxigênio no músculo — mesmo sem doença coronariana aterosclerótica significativa. É por isso que muitos pacientes com CMH se queixam de dor no peito típica de angina durante esforços, mesmo sem obstrução das coronárias epicárdicas.
Fibrose miocárdica: Com o passar dos anos, as áreas submetidas a estresse mecânico excessivo e isquemia repetida vão sendo substituídas por tecido fibroso, cicatricial. Essa fibrose é clinicamente relevante por dois motivos: ela reduz ainda mais a capacidade de contração e relaxamento do músculo, e ela cria substratos para arritmias graves, especialmente a taquicardia ventricular e a fibrilação ventricular.
Arritmias: Este é, sem dúvida, o aspecto mais temido da CMH. O músculo hipertrofiado, isquêmico e fibrótico é eletricamente instável. Pode gerar focos de atividade elétrica anormal que desencadeiam arritmias potencialmente fatais. A fibrilação atrial é comum — presente em até 20% a 25% dos pacientes ao longo da vida — e aumenta significativamente o risco de acidente vascular cerebral. Já as arritmias ventriculares malignas, como a fibrilação ventricular, são a causa da morte súbita cardíaca que caracteriza a face mais cruel e inesperada da CMH.
A face mais temida: morte súbita em jovens
A cardiomiopatia hipertrófica é, nos países ocidentais, a causa mais comum de morte súbita cardíaca em pessoas com menos de 35 anos, especialmente em atletas jovens e aparentemente saudáveis. Essa é a estatística que mais choca e que mais mobiliza a comunidade médica e os formuladores de políticas de saúde pública.
A imagem do atleta jovem, saudável, que desaba em campo durante uma partida de futebol ou no meio de uma corrida e não resiste é dolorosamente familiar. Nos Estados Unidos, estima-se que a CMH responda por cerca de 36% das mortes súbitas em atletas jovens. Casos emblemáticos de jogadores de basquete, futebol americano e outros esportes de alto rendimento que morreram subitamente por CMH sensibilizaram o público e ajudaram a colocar a doença em pauta.
O mecanismo é o seguinte: durante o exercício intenso, o coração bate mais rápido, o fluxo sanguíneo aumenta, a obstrução pode se acentuar, e a demanda elétrica sobre um miocárdio já instável pode ultrapassar o limiar crítico para o desencadeamento de uma arritmia fatal. Em segundos, a fibrilação ventricular leva à perda de consciência e, sem ressuscitação imediata, à morte.
A tragédia reside no fato de que muitas dessas vítimas não tinham qualquer sintoma prévio. A morte súbita pode ser a primeira — e última — manifestação da doença. Daí a importância dos programas de rastreamento cardiovascular em atletas, amplamente discutidos e implementados em diferentes países com resultados variados, mas com um consenso crescente de que o eletrocardiograma e o ecocardiograma devem fazer parte da avaliação pré-participação esportiva.
Em que idade a doença se torna mais agressiva?
A cardiomiopatia hipertrófica pode se manifestar em qualquer fase da vida, desde a infância até a velhice. Contudo, a expressão clínica da doença segue uma trajetória que os especialistas já conhecem bem.
O período de maior risco para morte súbita é a adolescência e o início da vida adulta — entre os 10 e os 35 anos. Essa janela de vulnerabilidade coincide com o pico de atividade física intensa, seja no contexto esportivo competitivo, seja em atividades recreativas vigorosas. O coração jovem do portador de CMH está sujeito a demandas metabólicas elevadas sobre uma estrutura já comprometida geneticamente.
A hipertrofia em si tende a ser mais pronunciada durante os anos de crescimento. Bebês e crianças pequenas raramente apresentam CMH grave, pois o espessamento é um processo progressivo que acompanha o desenvolvimento somático. É durante a puberdade, com a ação dos hormônios anabólicos e o crescimento acelerado, que o espessamento ventricular frequentemente se torna mais evidente e sintomático.
Na faixa dos 30 aos 60 anos, a doença entra em uma fase diferente. O risco de morte súbita diminui ligeiramente em termos relativos, mas outros problemas ganham protagonismo: a obstrução da via de saída se torna mais persistente, a fibrilação atrial é cada vez mais frequente, e os sintomas de insuficiência cardíaca — falta de ar, edema, limitação ao exercício — passam a dominar o quadro clínico de uma parcela crescente dos pacientes.
Em pacientes com mais de 60 ou 70 anos, a CMH pode evoluir para uma fase chamada de "queimada" ou "end-stage", em que a hipertrofia intensa ao longo de décadas leva ao esgotamento do músculo cardíaco. O ventrículo esquerdo, que antes era pequeno e hipercontrátil, pode dilatar-se e perder função sistólica — tornando-se semelhante, nesse estágio terminal, a uma cardiomiopatia dilatada. Essa evolução ocorre em uma minoria dos pacientes (menos de 5%), mas representa um dos desfechos mais graves da doença.
Os sintomas que não devem ser ignorados
A CMH é muitas vezes apelidada de "doença do silêncio" porque grande parte dos portadores não tem qualquer sintoma por anos ou décadas. Quando os sintomas aparecem, eles podem ser facilmente confundidos com outras condições mais banais.
Os principais sintomas incluem:
Dispneia aos esforços — falta de ar desproporcionalmente intensa para o nível de atividade realizado. Subir um lance de escadas, caminhar em ritmo acelerado ou praticar qualquer exercício moderado pode desencadear sensação de sufocamento. Isso ocorre porque o ventrículo rígido e obstruído não consegue aumentar adequadamente o débito cardíaco conforme a demanda do organismo.
Síncope e pré-síncope — episódios de desmaio ou quase-desmaio, especialmente durante ou imediatamente após exercícios. A síncope de esforço é um sinal de alarme que deve ser investigado imediatamente, pois pode preceder episódios de arritmia fatal.
Dor torácica — sensação de aperto, pressão ou queimação no peito, frequentemente desencadeada por esforço. Como mencionado, ocorre por isquemia do músculo espessado, independentemente de doença coronariana.
Palpitações — sensação de batimentos irregulares, acelerados ou "saltos" no ritmo cardíaco. Podem corresponder a extrassístoles benignas ou a arritmias mais significativas, como fibrilação atrial ou episódios de taquicardia ventricular.
Intolerância ao exercício — progressiva incapacidade de realizar atividades físicas que antes eram executadas sem dificuldade. Muitos pacientes vão se adaptando imperceptivelmente, reduzindo o nível de atividade ao longo dos anos, sem perceber que estão compensando uma limitação cardíaca real.
É fundamental destacar que qualquer combinação desses sintomas em uma pessoa jovem — especialmente com história familiar de morte súbita cardíaca, cardiomiopatia ou "doença do coração" em parentes de primeiro grau — deve motivar investigação cardiológica completa. A história familiar é, muitas vezes, o elo que falta para o diagnóstico.
Como a doença é identificada: os exames fundamentais
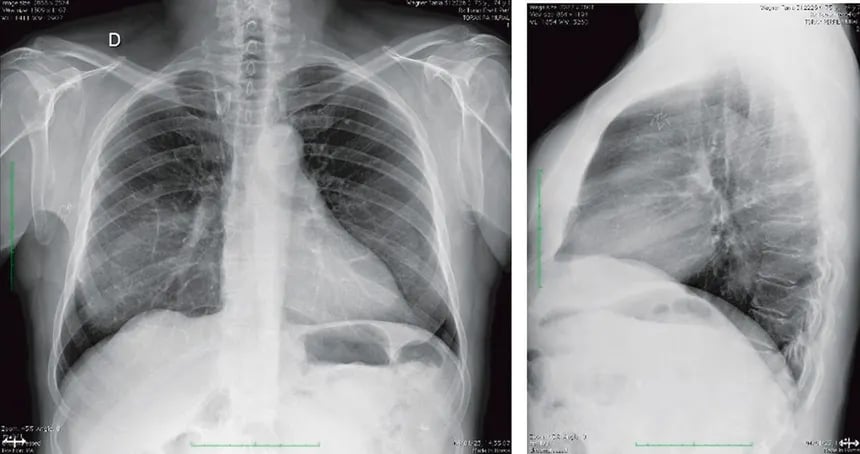
O diagnóstico da cardiomiopatia hipertrófica é essencialmente clínico e por imagem, apoiado pela genética molecular quando disponível. Nenhum exame laboratorial de sangue rotineiro — colesterol, hemograma, glicemia — detecta a CMH. É necessário investigar o coração de forma direta.
Ecocardiograma transtorácico
O ecocardiograma é o pilar diagnóstico da CMH. Por meio de ondas de ultrassom, ele permite visualizar o coração em tempo real, medir a espessura das paredes do ventrículo esquerdo, avaliar a função sistólica e diastólica, identificar a presença de obstrução da via de saída e detectar o movimento anormal da valva mitral (SAM). O critério ecocardiográfico mais utilizado para o diagnóstico de CMH é uma espessura do septo interventricular ou da parede posterior igual ou superior a 15 milímetros na ausência de outra causa que justifique a hipertrofia — ou 13 milímetros quando há história familiar de CMH ou mutação genética conhecida.
O ecocardiograma com Doppler colorido e espectral fornece informações adicionais sobre gradientes de pressão na via de saída do ventrículo esquerdo, avalia a valva mitral e mensura o grau de insuficiência mitral frequentemente associada à obstrução.
Eletrocardiograma (ECG)
O eletrocardiograma de repouso é anormal em mais de 90% dos pacientes com CMH sintomática. As alterações mais comuns incluem sinais de hipertrofia ventricular esquerda, ondas Q anormais (especialmente em derivações inferiores e laterais), alterações de repolarização com inversão de onda T, bloqueios de ramo e, em alguns casos, padrão de pré-excitação ventricular associado à síndrome de Wolff-Parkinson-White.
O ECG é especialmente útil no rastreamento de familiares de primeiro grau de portadores de CMH: mesmo antes de qualquer espessamento visível ao ecocardiograma, o eletrocardiograma pode mostrar alterações sugestivas da doença.
Holter de 24 horas
O monitoramento contínuo do ritmo cardíaco por 24 horas (Holter) ou períodos mais longos é fundamental para identificar arritmias que não aparecem no ECG de repouso. Episódios de taquicardia ventricular não sustentada (TVNS) — séries de três ou mais batimentos ventriculares anormais, com duração inferior a 30 segundos — são achados importantes no Holter de pacientes com CMH e integram os critérios de estratificação de risco para morte súbita.
Teste ergométrico e ergoespirometria
O teste de esforço em esteira ou bicicleta permite avaliar a resposta do coração ao exercício, identificar arritmias induzidas por esforço, medir a variação do gradiente obstrutivo com o exercício e, por meio da ergoespirometria (análise dos gases expirados durante o esforço), quantificar com precisão a capacidade funcional do paciente. Uma queda ou resposta plana da pressão arterial durante o exercício é sinal de risco aumentado para eventos adversos.
Ressonância magnética cardíaca (RMC)
A ressonância magnética cardíaca representa o mais poderoso instrumento diagnóstico e prognóstico disponível atualmente para a CMH. Sua resolução espacial superior ao ecocardiograma permite identificar padrões de hipertrofia que às vezes escapam ao ultrassom — especialmente nas regiões apical e lateral do ventrículo esquerdo, zonas tecnicamente difíceis de avaliar pela ecocardiografia convencional.
O ponto de destaque absoluto da RMC é a capacidade de detectar fibrose miocárdica por meio da técnica de realce tardio de gadolínio (Late Gadolinium Enhancement — LGE). A presença e a extensão da fibrose são preditores independentes de eventos adversos, incluindo morte súbita, progressão para insuficiência cardíaca e arritmias. Pacientes com fibrose extensa (superior a 15% da massa miocárdica) têm risco significativamente aumentado e podem se beneficiar de implante de cardioversor-desfibrilador implantável (CDI) como prevenção primária.
A RMC cardíaca está atualmente indicada em todos os pacientes com suspeita ou diagnóstico confirmado de CMH, idealmente antes de qualquer decisão terapêutica importante.
Teste genético
O teste genético para CMH envolve o sequenciamento de um painel de genes sarcoméricos associados à doença. Sua principal utilidade está no rastreamento em cascata de familiares — uma vez identificada a mutação no paciente-índice, é possível testar os parentes de primeiro grau e identificar os portadores antes mesmo de qualquer manifestação clínica ou ecocardiográfica.
O teste genético positivo não altera, por si só, o tratamento da CMH, mas tem implicações profundas para o aconselhamento genético, planejamento familiar e monitoramento de portadores assintomáticos. O teste negativo, por outro lado, não exclui o diagnóstico — cerca de 30% a 40% dos pacientes com CMH clinicamente definida não têm mutação identificada pelos painéis disponíveis, o que pode significar mutações em genes ainda não catalogados ou variantes de significado incerto.
Cateterismo cardíaco
O cateterismo é reservado para situações específicas: confirmação hemodinâmica de obstrução grave antes de procedimentos invasivos (miomectomia cirúrgica ou ablação septal por álcool), avaliação de doença coronariana concomitante em pacientes com fatores de risco, ou investigação de pressões de enchimento em casos de insuficiência cardíaca avançada. Não é um exame de primeira linha no diagnóstico da CMH.
Estratificação de risco: quem tem maior perigo?
Nem todo portador de CMH tem o mesmo risco de morte súbita. A estratificação de risco é uma das atividades mais importantes — e mais desafiadoras — no acompanhamento desses pacientes.
Os principais fatores de risco para morte súbita em CMH incluem: história pessoal de parada cardíaca ou arritmias ventriculares sustentadas; síncope inexplicada recente; hipertrofia ventricular esquerda maciça (espessura ≥ 30 mm); história familiar de morte súbita em parentes jovens; TVNS ao Holter; resposta pressórica anormal ao exercício; e fibrose miocárdica extensa à ressonância magnética.
A presença de múltiplos fatores de risco aumenta substancialmente a probabilidade de eventos adversos. Com base nessa estratificação, os cardiologistas decidem pela indicação de cardioversor-desfibrilador implantável (CDI) — dispositivo que monitora o ritmo cardíaco continuamente e aplica um choque elétrico interno para reverter uma fibrilação ventricular caso ela ocorra.
Tratamento: controlar, não curar
A cardiomiopatia hipertrófica não tem cura definitiva disponível na maioria dos casos. O tratamento atual tem como objetivos aliviar os sintomas, melhorar a qualidade de vida, prevenir complicações e reduzir o risco de morte súbita.
Tratamento medicamentoso
Os betabloqueadores são os medicamentos de primeira linha para pacientes com CMH obstrutiva e sintomática. Eles reduzem a frequência cardíaca, diminuem a força de contração e, com isso, reduzem o gradiente obstrutivo e melhoram o enchimento ventricular. Propranolol, metoprolol e atenolol são os mais utilizados.
Os bloqueadores dos canais de cálcio não diidropiridínicos, como o verapamil e o diltiazem, são alternativas para pacientes que não toleram ou não respondem aos betabloqueadores. Eles também reduzem a frequência cardíaca e melhoram o relaxamento diastólico.
A disopiramida, um antiarrítmico com propriedades inotrópicas negativas, pode ser associada aos betabloqueadores em casos de obstrução grave refratária.
Em 2022, uma nova classe de medicamento foi aprovada para o tratamento da CMH obstrutiva: o mavacamten, um inibidor seletivo da miosina cardíaca que age diretamente no mecanismo de contração do sarcômero, reduzindo a hipercontratilidade característica da doença. Os estudos clínicos demonstraram reduções significativas no gradiente obstrutivo e melhora nos sintomas, representando o primeiro avanço terapêutico verdadeiramente mecanístico — voltado para a causa, não apenas para os sintomas — em décadas.
Procedimentos invasivos
Quando o tratamento medicamentoso não é suficiente para controlar os sintomas em pacientes com obstrução grave, dois procedimentos invasivos estão disponíveis:
A miomectomia septal cirúrgica (operação de Morrow) é o procedimento padrão-ouro. Consiste na remoção cirúrgica de uma porção do septo interventricular hipertrofiado, aliviando a obstrução. Realizada por cirurgiões experientes em centros especializados, apresenta taxas de sucesso superiores a 90% e mortalidade operatória inferior a 1%.
A ablação septal por álcool é uma alternativa percutânea (feita por cateter, sem cirurgia aberta) em que pequena quantidade de álcool é injetada em uma artéria septal, provocando um infarto localizado e controlado do septo hipertrofiado. É especialmente indicada para pacientes de maior risco cirúrgico.
Cardioversor-desfibrilador implantável (CDI)
Para os pacientes classificados como de alto risco para morte súbita, o implante de um CDI é a medida mais eficaz de prevenção. O dispositivo, implantado subcutaneamente ou sob a clavícula, monitora continuamente o ritmo cardíaco e está pronto para aplicar um choque elétrico interno sempre que detectar uma arritmia fatal — literalmente salvando a vida do portador.
Prevenção: o que é possível fazer?
Como a CMH é uma doença genética, não é possível "prevenir" que o gene mutado cause espessamento do músculo cardíaco. No entanto, várias medidas são eficazes para reduzir o risco de complicações, prevenir a morte súbita e identificar portadores antes que a doença se manifeste de forma grave.
Rastreamento familiar em cascata
Toda pessoa diagnosticada com CMH deve orientar seus familiares de primeiro grau — pais, filhos, irmãos — a procurar avaliação cardiológica com ECG e ecocardiograma. Se disponível, o teste genético direciona ainda mais o rastreamento. Identificar um portador precocemente significa monitorá-lo adequadamente, restringir atividades de risco quando necessário e tratar antes de uma complicação grave.
Restrição de esportes competitivos de alta intensidade
Para portadores com CMH confirmada, especialmente aqueles com obstrução, hipertrofia maciça ou arritmias documentadas, a prática de esportes de alta competição geralmente é contraindicada pelas diretrizes cardiológicas internacionais. Isso não significa que o portador não possa se exercitar — atividades moderadas e supervisionadas são encorajadas e benéficas — mas os esportes de competição com exigência máxima e prolongada do sistema cardiovascular representam risco aumentado.
Evitar medicamentos e situações que piorem a obstrução
Certos medicamentos e situações agravam a obstrução na CMH: diuréticos em excesso (que reduzem o volume do ventrículo), vasodilatadores puros, nitratos, digitálicos em altas doses e desidratação intensa. Portadores devem ser orientados sobre essas interações e informar sempre o diagnóstico ao receberem qualquer prescrição médica.
Controle da fibrilação atrial
A prevenção e o tratamento agressivo da fibrilação atrial é outra frente importante. Pacientes com CMH e FA têm risco elevado de AVC tromboembólico, e a anticoagulação é indicada na maioria dos casos, independentemente do escore de risco convencional. O controle do ritmo e da frequência cardíaca na FA também é fundamental para manter a estabilidade clínica.
Acompanhamento periódico em centro especializado
O acompanhamento regular — com reavaliação clínica, ecocardiograma periódico, Holter anual e ressonância magnética quando indicada — é fundamental para detectar precocemente mudanças no padrão da doença, reavaliar o risco de morte súbita e ajustar o tratamento conforme a evolução.
A vida com cardiomiopatia hipertrófica: entre o medo e a esperança
Receber o diagnóstico de cardiomiopatia hipertrófica pode ser um momento de profundo impacto emocional. A palavra "cardiomiopatia" carrega peso. A associação com morte súbita assusta. O fato de ser uma doença genética levanta questões sobre filhos, sobre herança, sobre o futuro.
Mas a narrativa contemporânea da CMH é, ao contrário do que se poderia imaginar, majoritariamente otimista.
A grande maioria dos portadores de CMH — estudos consistentes indicam acima de 80% — tem prognóstico favorável quando acompanhados adequadamente. A expectativa de vida de pacientes com CMH seguidos em centros especializados é próxima à da população geral da mesma faixa etária. As ferramentas diagnósticas e terapêuticas disponíveis hoje — da ressonância magnética ao CDI, do mavacamten à miomectomia — transformaram completamente o panorama da doença nas últimas duas décadas.
O maior inimigo da CMH não é a doença em si, mas o desconhecimento. O portador que não sabe que tem a condição é aquele que pratica esportes de alta intensidade sem restrições, que ignora sintomas de alarme, que não alerta os familiares. É por isso que a educação médica e a conscientização pública são, em última análise, as ferramentas mais poderosas de prevenção disponíveis.
A cardiomiopatia hipertrófica não define uma sentença. Define uma condição que precisa ser conhecida, monitorada e manejada — para que quem a carrega no peito possa viver com plenitude, segurança e, sobretudo, informação.
Escrito por: Equipe Editorial Saldo e Vida Conteúdo focado em transparência financeira e bem-estar integral.
Contato
Fale conosco para sugestões e dúvidas
saldoevida@saldoevida.com
© 2025. All rights reserved.
Sobre Nós